Theory of Edman Sequencing
An overview of Protein Sequencing using Edman Degradation and Automation using Shimadzu PPSQ Instruments.
Introduction of Edman Degradation
Proteins are molecules consisting of monomers called amino acids that are linked together covalently by amide bonds between the C-terminus of one amino acid and N-terminus of another amino acid1 (figure 1). The number of amino acid monomers can vary greatly depending on the function of the peptide or protein of interest. Determination of the amino acid sequence of peptides and proteins has been an area of interest to aid in the understanding of the structure2,3, and function4 of these biomolecules. Multiple methods of peptide and protein sequencing5 have been used to determine the sequence of key peptides and proteins; below we will discuss the method of Edman degradation in detail.
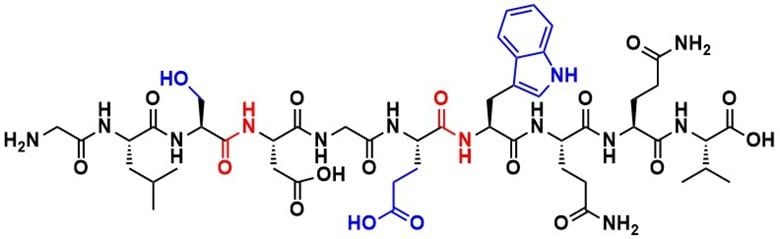
Figure 1. Structure of a polypeptide chain from the protein apomyoglobin from equine skeletal muscle, highlighting the amide bonds in red at the connection between apartic acid and serine, and glutamic acid and tryptophan. Amino acid side chains are for serine, glutamic acid and tryptophan are shown in blue. The sequence the shown peptide is NH2-Gly-Leu-Ser-Asp-Gly-Trp-Gln-Gln-Val-COOH (GLSDGWQQV).
Pehr Edman reported a method of amino acid sequencing in 19506, using phenyl isothiocyanate (PITC) to functionalize N-terminal amino acids from a peptide or protein of interest to aid in monomer detection. From this came the technique known as Edman degradation, a method for N-terminal sequencing of peptides and proteins. This method functionalizes the N-terminal amino acid for cleavage, leaving the remaining peptide or protein untouched (figure 2). The use of phenyl isothiocyanate provides a way to differentiate and detect amino acids using paper chromatography, UV detection or other methods through the addition of an aromatic ring. This process is done using PITC which forms a phenylthiocarbamoyl derivative with the N-terminus of the peptide under alkaline conditions. The peptide can then be cleaved under acidic conditions, creating a phenylthiodantoin (PTH) amino acid residue (scheme 1,2). Edman degradation can be performed repeatedly to cleave each sequential amino acid from the peptide or protein to obtain a full sequence.
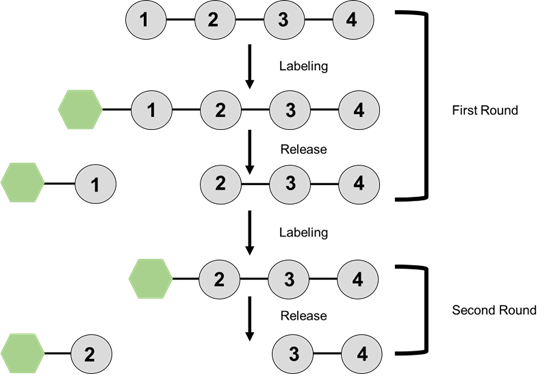
Figure 2. General schematic of the process of Edman degradation. Free N-terminal amino acids are labeled and cleaved from the main group. This process is repeated for each additional amino acid in the chain. Inspired by a figure in Biochemistry Berg, et al.7
Edman degradation provides a reliable method of single amino acid sequencing as other researchers at the time relied on the acidic hydrolysis of amide bonds to break down the peptide into smaller di- or tripeptides for sequencing. In some methods, such as those by Sanger8,9,10 and Consden11,12 partial acid hydrolysis of peptides was performed to determine sequences of smaller fragments by paper chromatography. Sanger utilized this method to determine the sequence of insulin, which contains a complex disulfide bonded structure. The method of generating differing size peptide fragments led to the proteomics technique of Peptide Mass Fingerprinting (PMF)13. The sequence of larger proteins could be identified by different mass spectrometry techniques (ex. LCMS, MALDI-TOF) of the smaller peptide fragments formed from acid hydrolysis or enzyme cleavage.
Edman continued to improve the degradation protocol. The first follow up report focused on streamlining the process of identifying the liberated PTH functionalized amino acid14. To determine the identity of cleaved and functionalized amino acids, Edman synthesized PTH functionalized amino acids to serve as standards for paper chromatography analysis. Column chromatography combined with light absorption was utilized to separate and detect some of the PTH amino acid adducts. In his work, he noted that the initial coupling reaction happened quickly between PITC and the amino acid, while conversion to the PTH amino acid took much longer. After reaction with PITC, the resulting 5-thiazolinone (ATZ) amino acid can be separated from the remaining protein and transferred for conversion to the PTH amino acid. This separation step allowed for minimal peptide hydrolysis during the acidic cleavage step of the functionalized amino acid.
Edman found that the process of forming a PTH amino acid broke down into three steps; (1) Coupling with PITC to from a phenylthiocarbamoyl derivative, (2) cleavage of the phenylthiocarbamoyl derivative in aqueous acid, forming the ATZ peptide adduct, (3) conversion of the ATZ peptide adduct into corresponding PTH amino acid. This would lead to the foundation of the automated Edman degradation sequenator. Automation of Edman degradation did not take long after Edman had reported the manual process. In a follow up publication, Edman and Begg detailed an apparatus setup of their “sequenator”15. The first reported sequenator could perform 15.4 degradation cycles in 24 hours, needing approximately 25 µmols of protein for sequencing. Their initial model peptide was apomyoglobin from the humpback whale, where they were able to sequence the first 60 amino acids from the N-terminal end. The sequenator had the ability to generate the PTH amino acids but required manual spotting on a thin layer chromatography (TLC) plate for analysis. The need for manual analysis made it challenging to determine the effectiveness of the reaction until much later, potentially wasting time and resources16. The origination of the sequenator brought upon the opportunity to further develop the automated system.
There are few limitations for Edman degradation; first being that there cannot be any N-terminal modifications on the peptide. Any changes at this position will prevent the coupling of PITC and thus prevent formation of a PTH amino acid. In the early stages of development of Edman degradation, Edman found that some proteins became insoluble after a few rounds of sequencing. The oxidation of disulfide bonds prior to sequencing resolved this issue, making reduction of disulfide bonds a necessary pretreatment step for efficient sequencing.
In the original sequenator, the peptide was spin coated on the walls of the reaction chamber. Now, peptides and proteins are bound to a membrane for analysis. The peptides for Edman degradation can come from a mixture of different peptides and proteins. This mixture can be separated by size using SDS-polyacrylamide gel electrophoresis (PAGE) gel and transferred to a Polyvinylidene difluoride (PVDF) membrane through electroblotting17 . PVDF membranes are commonly used in western blotting applications where it facilitates the transfer of a peptide from a gel to the membrane for subsequent analysis. The introduction of electroblotting onto PVDF membranes from SDS-PAGE gels allowed for an additional use of Edman degradation. Peptides and proteins bind to PVDF membranes through hydrophobic and dipole interactions. PVDF is a durable membrane, making it suitable for sequencing reactions with multiple washing steps as it will hold up better than a nitrocellulose membrane that can also be used for electroblotting. Another membrane that can be used for automated Edman degradation is a siliconized-glass fiber disc. Modified glass-fiber sheets are prepared through the reaction on the surface of poly(methyl-3,3,3-trifluoropropylsiloxane) in trifluoroacetic acid18. Glass-fiber discs (GFD) exhibit a higher protein-binding capacity, proving to be beneficial for the sequencing of lower quantities of proteins. The membrane choice for automated Edman degradation will depend on the peptide or protein that you are analyzing and its ability to bind to the membrane.
Automation of Edman Degradation
Since the first reported automated Edman degradation sequenator, others joined in to continually simplify and streamline the process. The use of high-performance liquid chromatography (HPLC) had been implemented to aid in the separation and detection of PTH amino acids. The use of reverse phase HPLC, where the stationary phase is non-polar and the mobile phase is polar, allowed for a mixture of PTH amino acids to be separated and serve as a standard for unknown PTH amino acid detection19. The use of RP-HPLC was the final step towards full automation of Edman degradation. Now samples can be directly injected into the HPLC system to allow for continual analysis and troubleshooting as early as needed.
Shimadzu Scientific Instruments entered the automated Edman degradation field with the introduction of the PPSQ, a system that automates Edman degradation and analysis through connection with an HPLC system for detection of PTH amino acids. The Shimadzu PPSQ system automates Edman degradation in the gas phase to sequence proteins from the N-terminus. The following sequence is shown in Scheme 1 and explained here. The protein sample is coupled at the N-terminus with 5% Phenylisothiocyanate (PITC) under basic conditions created by 30% Methylpiperidine for gradient analysis conditions or 12% Trimethylamine for isocratic analysis. The excess reagents are then removed by ethyl acetate, and under acidic conditions from Trifluoroacetic acid, the Phenylthiocarbamoyl derivative is cleaved from the remaining protein. 1-Chlorobutane is then used to extract the derivative from the reactor. The cleaved derivative is then converted with 25% Trifluoroacetic acid into a Phenylthiohydantoin (PTH) amino acid derivative. With a reaction time less than 50 minutes, the system derivatizes the primary amino acids of a protein from the N-terminal, cleaves the derivatives at the amide bond, and injects the PTH amino acid residue into an HPLC. The amino acid is then identified by comparing the retention time of residue to the retention times of PTH amino acids in a standard solution. This process can be repeated to detect up to 30 PTH amino acids (in ideal sample conditions).
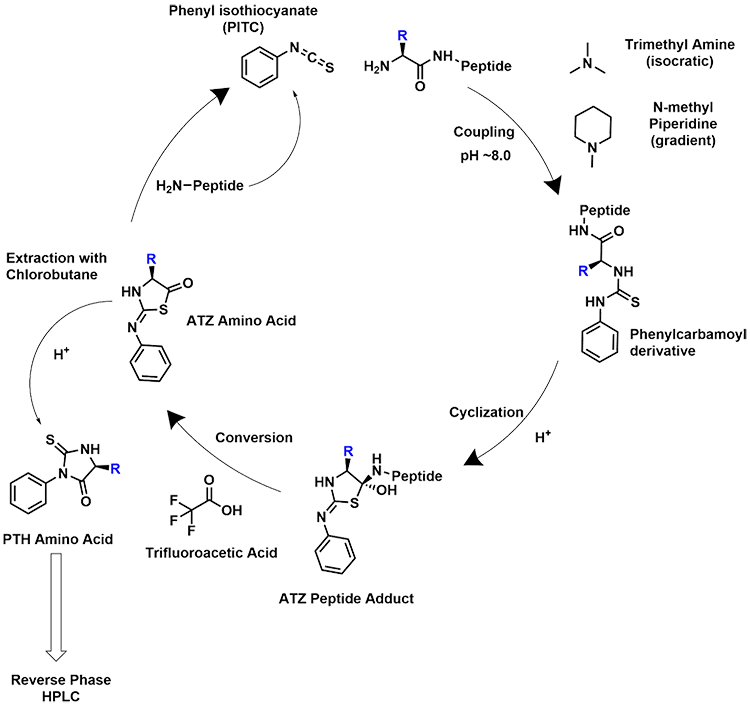
Scheme 1. Edman degradation cycle, amino acid side chain is denoted as “R”.
A proposed mechanism for the formation of PTH amino acids is shown in Scheme 2. The initial coupling of PITC to the free N-terminus occurs through a nucleophilic attack of the carbon of the isothiocyanate of PITC using the free amine of the peptide of interest. After the initial coupling, a cyclization occurs allowing for the generation of a tetrahedral center with the remainder of the peptide being part of it. Through a proton exchange, the remaining peptide is released and the currently analyzed amino acid as an ATZ amino acid. At this point the ATZ amino acid undergoes rearrangement to form the PTH amino acid.
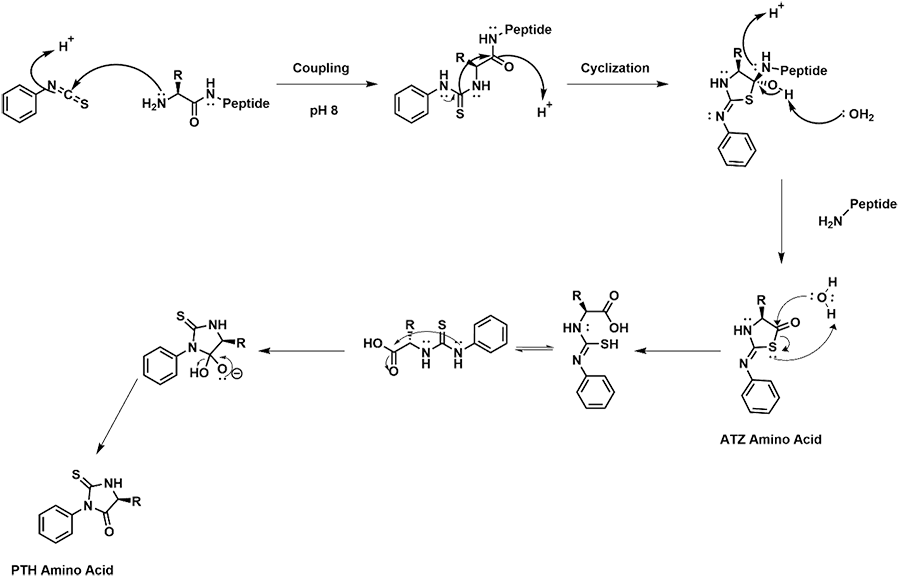
Scheme 2. Mechanism for the coupling of the N-terminal amino acid of a peptide, and conversion of the amino acid with PITC to a PTH amino acid for detection.
During HPLC analysis of the PTH amino acid, there are some reaction byproducts that are seen in the resulting chromatogram. Two major byproducts detected are DPTU and DPU, both are a result of PITC side reactions. The proposed mechanism for their formation is shown in scheme 3. The use of anhydrous solvents and nitrogen atmosphere can reduce the amount of these byproducts seen in the chromatogram.
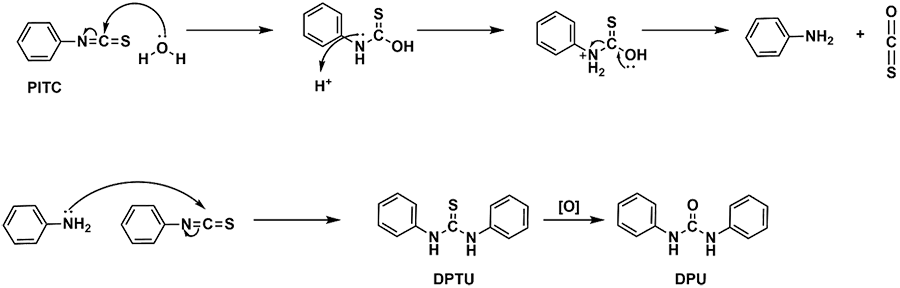
Scheme 3. Mechanism for the formation of two by-products commonly seen in the HPLC chromatograms. DPTU (diphenylthiourea) is formed from the condensation of PTIC molecules. DPU (diphenylurea) is the oxidized version of DPTU.
[1] Berg, J.M., Tymoczko, J.L., Gatto, G.J., Stryer, L. Protein Composition and Structure. In Biochemistry, 8th ed.; W.H. Freeman & Company, 2015; pp 27-39.
[2] Pauling, L., Corey, R. B., Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA 1951, 37 (4), 205-211. DOI: 10.1073/pnas.37.4.205
[3] Pauling, L., Corey, R.B., Branson, H.R. The Pleated Sheet, A New Layer Configuration of Polypeptide Chains. Proc Natl Acad Sci USA 1951, 37(5), 251-256. DOI: 10.1073/pnas.37.5.251
[4] Eisenberg, D. The discovery of the α-helix and β-sheet, the principal structural features of proteins. Proc Natl Acad Sci USA 2003, 100 (20), 11207-11210. DOI: 10.1073/pnas.2034522100
[5] Zhang, G., Annan, R.S., Carr, S.A., Neubert, T.A. Overview of Peptide and Protein Analysis by Mass Spectromerty. Curr Protoc Mol Biol. 2014, 108 (1), 10.21.1-10.21-30. DOI: 10.1002/0471140864.ps1601s62
[6] Edman, P. Method for Determination of the Amino Acid Sequence in Peptides. Acta Chem. Scand. 1950, 4 283-293. DOI: 10.3891/acta.chem.scand.04-0283
[7] Berg, J.M., Tymoczko, J.L., Gatto, G.J., Stryer, L. Mass Spectrometry. In Biochemistry, 8th ed.; W.H. Freeman & Company, 2015; pp 87.
[8] Sanger, F. The free amino groups of insulin. Biochem J 1945, 39 (5), 507-515. DOI: 10.1042/bj0390507
[9] Sanger, F. Oxidation of Insulin by Performic Acid. Nature 1947, 160, 295-296.
[10] Sanger, F. Some Peptides from Insulin. Nature 1948, 162, 491-492.
[11] Consden, R., Gordon, A.H., Martin, A.J.P. The identification of lower peptides in complex mixtures. Biochem J. 1947, 41 (4), 590-596. DOI: 10.1042/bj0410590fryan
[12] Consden, R., Gordon, A.H., Martin, A.J.P. and Synge, R. Biochem J. 1947, 41, 596-602. DOI: 10.1042/bj0410596
[13] Henzel, W.J., Watanabe, C., Stults, J.T. Protein identification: the origins of peptide mass fingerprinting. JASMS 2003, 14 (9), 931-942. DOI: 10.1016/S1044-0305(03)00214-9
[14] Edman, P. Phenylthiohydantoins in Protein Analysis. Ann. N.Y. Acad. Sci. 2006, 602-610. DOI: 10.1111/j.1749-6632.1960.tb20056.x
[15] Edman, P., Begg, G. A Protein Sequencer. Euro. J. Biochem. 1967, 1, 80-91. DOI: 10.1111/j.1432-1033.1967.tb00047.x
[16] Niall, H.D. Automated Edman Degradation: The Protein Sequinator. Meth.Enzymol. 1973, 6879 (73), 27039-8. DOI: 10.1016/s0076-6879(73)27039-8
[17] Komatsu, S. Western Blotting Using PVDF Membranes and Its Downstream Applications. In Western Blotting ; Springer Protocols, 2015; pp 227-236. DOI: 10.1007/978-1-4939-2694-7_24
[18] Eckerskorn, C., Mewes, W., Goretzki, H., Lottspeich, F. A new siliconized-glass fiber as support for protein-chemical analysis of electroblotted proteins. Eur. J. Biochem. 1988, 176, 509-519. DOI: 10.1111/j.1432-1033.1985.tb09157.x
[19] Knecht, R., Seemuller, U.,Liersch, M., Fritz, H., Braun, D.G., Chang, J.Y. Sequence Determination of Eglin C Using Combined Microtechniques of Amino Acid Analysis, Peptide Isolation and Automatic Edman Degradation. Anal. Biochem. 1983, 130, 65-71. DOI: 10.1016/0003-2697(83)90650-4