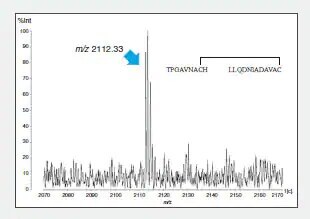
Fig. 3 MS Spectrum of Thermolysin Digest of Peptide Fragment A (m/z 3636.36)
If the gene sequence that codes for the target antibody indicates a cysteine residue, the Guidelines require the determination of the number and positions of all free sulfhydryl groups and disulfide bonds. This is evaluated using peptide mapping (under reducing conditions and non-reducing conditions), mass spectrometry or another suitable method.
Introduced here is the analysis using a combination of peptide mapping and mass spectrometry. This method involves digesting the target protein in a proteolytic enzyme such as trypsin under various conditions and comparing the molecular weights of the generated peptide fragments with the theoretical values. The test procedures and results for the analysis of human lysozyme are introduced.
Fig. 1 shows the amino acid sequence and predicted S-S bonds (indicated by straight lines) of the human lysozyme synthesized in the test tube. Table 1 shows the theoretical values and MS measured values for the trypsin-digested peptide fragments (associated with S-S bonds).
Fig. 2 shows part of the MS spectrum of the trypsin-digested peptide fragments.
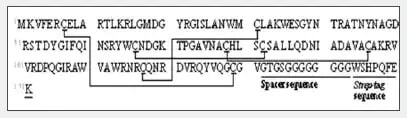
Fig. 1 Amino Acid Sequence and Predicted S-S Bonds of the Synthesized Human Lysozyme
The m/z 3239.22 ion disappears due to reduction and alkylation of peptide fragment B, indicating that peptide 3 and peptide 4 form intramolecular S-S bonds (Fig. 2). The same method confirmed intramolecular S-S bonds in peptide fragment C. (Data omitted.)
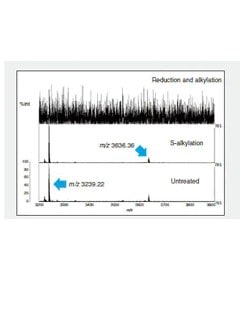
Fig. 2 MS Spectrum of Recovered Peptide Fragments (m/z 3200 to m/z 3900)
Comparison of the reduction/alkylation and untreated MS spectra for peptide fragment A indicates that peptide 1 and peptide 2 form intramolecular S-S bonds. However, the combination with the cysteine residue cannot be determined from the results in Fig. 2 only.
To confirm this combination, the peptide 1 intramolecular S-S bonds were identified using LC to separate and purify peptide fragment A, digestion in another protease (thermolysin), and determination of the mass of the generated fragments (Fig. 3).
Fig. 3 MS Spectrum of Thermolysin Digest of Peptide Fragment A (m/z 3636.36)
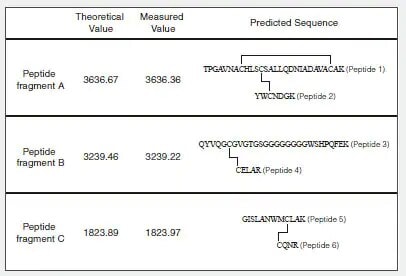
Table 1 Theoretical Values and Measured Values for Predicted Peptide Fragments (Associated with S-S Bonds)

The AXIMA Performance is one of the most powerful tools in mass spectrometry, delivering information-rich spectra with greater sensitivity and higher confidence in identification. It is an extremely versatile and powerful TOF-TOF system, integrating workflows for a diverse range of analytical needs.
AAPS Pharm Sci 360 2026 (American Association of Pharmaceutical Scientists)
October 26-28
Ernest Morial Convention Center
New Orleans, LA
Booth #1204
ASMS (American Society for Mass Spectrometry) 2026
May 31 - June 4
San Diego Convention Center
San Diego, CA
Suite 5, 6
Pharma Community Network Event
Shimadzu Scientific Instruments and ZefSci invite you and your analytical teams to a pharma community networking event to celebrate 150 years of science and innovation at The Foundry & Lux in South San Francisco. Our event will be built around innovation, collaboration, and connection.
Shimadzu Scientific Instruments Opens Boston Location of Its R&D Center Focus will be on promoting customer-oriented development to expand business in the pharmaceutical field
Shimadzu Scientific Instruments, Inc. (SSI, Columbia, Maryland, USA), a Shimadzu Group company, has opened a satellite lab in Boston, Massachusetts to be the base of its collaborative research and development activities on the East Coast. Established to conduct research and development more closely linked to customers, SSI's R&D Center consists of three bases, with the main facilities at its Maryland headquarters, a West Coast location, and this new space in Boston near the city center. The Boston lab was set up by partnering with Labshares, a shared laboratory service provider for life science companies.